Understanding Dystonia: A Practical Approach to Diagnosis and Management
Clinical history helps corroborate the diagnosis of dystonia and provides clues to its etiology; however, the diagnosis rests on clinical examination, because no gold standard laboratory or imaging tests are available.

The term “dystonia” was first used in 1911 by Oppenheim,1 who described 4 individuals with “dystonia musculorum deformans” and proposed an organic etiology for this unusual disorder. Fahn et al2 defined dystonia as a syndrome of sustained muscle contractions, frequently causing twisting and repetitive movements or abnormal postures. Over time, multiple attempts to define and classify dystonia have been made.
When an individual presents with abnormal postures and involuntary movements, the first step is to establish whether the phenomenology is consistent with true dystonia. Clinical history helps corroborate the diagnosis of dystonia and provides clues to its etiology; the diagnosis rests on clinical examination, because no gold standard laboratory or imaging tests are available. Once true dystonia is confirmed, specific tests may be warranted to establish etiology.
In this review, we discuss the phenomenology, diagnosis, and treatment of dystonia; outline the diagnostic workup; and provide practical algorithms for the treating clinician.
Phenomenology of Dystonia
Dystonia is characterized by abnormal movements and postures. Three main points should be addressed to secure a diagnosis of dystonia and rule out a condition mimicking dystonia (called “pseudodystonia”):
1. What are the characteristics of the involuntary movements (eg, speed, amplitude, rhythm)?
2. What are the characteristics of the abnormal postures?
3. Are there ancillary features or diagnostic clues that support the diagnosis?
Dystonia is a syndrome that can arise from multiple etiologies. In its revised classification system, the Movement Disorders Society implemented a 2-axis classification approach: Axis I, based on history and clinical examination; and Axis II, grounded on pathogenesis.3 This classification has been updated recently.4
Clinical Characteristics (Axis I)
Considerations for the History and Physical Examination
Flowcharts outlining the diagnostic workup combining Axis I and Axis II are presented for adult-onset dystonia (Figure 1) and early-onset dystonia (Figure 2).

Figure 1. Diagnostic workup in adult-onset dystonia combining Axis I and Axis II. Abbreviations: CBS, corticobasal syndrome; CSF, cerebrospinal fluid; NBIA, neurodegeneration with brain iron accumulation; NGS, next-generation sequencing; WGS, whole-genome sequencing.

Figure 2. Diagnostic workup in early-onset dystonia combining Axis I and Axis II. Abbreviations: DRD, dopa-responsive dystoni; NBIAs, neurodegeneration brain iron accumulations.
Age at Onset. Age at onset offers an essential clue to etiology. Dystonia onset in childhood underpins a genetic etiology. Dystonia is one of the most common motor manifestations of cerebral palsy, a condition in which genetic background plays a significant role.5 Adult-onset focal or segmental dystonia is more likely sporadic, although some genetic dystonias may manifest later in life.
Family History. Family history should be investigated because dystonia may be inherited, and patterns of inheritance have been reported (Table 1). Over the past decade, with advances in genetic techniques, an increasing number of genes have been identified. A negative family history does not rule out genetic etiology; some genes display reduced penetrance, and individuals may have de novo sequence variations. Furthermore, the same variant may be associated with different clinical phenotypes. Focal dystonia may occur among individuals carrying defects in genes more commonly associated with generalized dystonia during childhood, such as the TOR1A gene at the DYT1 locus.6



Natural History. An acute presentation of dystonia hints at exposure to dopamine blocking agents, stroke, or a functional etiology. Rapid-onset dystonia-parkinsonism may also present acutely, and acute onset after metabolic decompensation may be seen in dystonia associated with inborn errors of metabolism.7 A subacute course developing over days or weeks typically implies an autoimmune etiology, whereas a chronic course evolving over months or years is typical of focal adult-onset dystonia. Symptom variability in a short timeframe may be seen in paroxysmal dystonia (ie, episodes of dystonia triggered by specific stimuli, with return to baseline after cessation of stimuli) or DOPA-responsive dystonia (ie, diurnal fluctuations, with symptoms worsening in the evening due to the circadian rhythm of dopamine metabolism).4
Characteristics of Dystonic Movements
Dystonic movements are the slowest involuntary hyperkinetic movements. They are usually patterned, twisting, and repetitive, and may have a superimposed tremor-like or jerky-like quality. The term “dystonic tremor” is currently discouraged, and tremor and dystonia should be described as two separate, but coexisting entities.3,4
Compared with dystonia, myoclonic movements are faster; choreic movements are typically random and flowing, and vary in duration, direction, and amplitude; tics are usually temporarily suppressible, preceded by a premonitory urge, and followed by a sensation of relief; and tremor is rhythmic and oscillatory.
Characteristics of Body Postures. Dystonic postures are associated with intermittent increases in tone. A fixed or sustained posture suggests spasticity; orthopedic problems, such as contractures; muscular deformities; or a functional etiology.6 Exceptions to this rule are corticobasal syndrome and rapid-onset dystonia parkinsonism.7,8
Ancillary Features or Diagnostic Clues
Task Specificity and Relationship with Voluntary Movements. Selective activation of dystonia during the performance of a specific task, such as playing a musical instrument or using a specific tool, is a unique feature of dystonia.9 Task complexity and precision likely increase the risk of developing task-specific dystonia (eg, guitarists and pianists are at higher risk among musicians; classical musicians are more at risk than rock or jazz musicians). Changes in the task routine are also a risk factor.
Dystonic Spread. Dystonia may spread in body distribution or task specificity. Dystonia may start in a focal distribution, affecting only one region (eg, eyes, neck, hand), and later become segmental or generalized. In addition, it may initially affect one task selectively, and later involve other tasks (eg, individuals with writer’s cramp developing dystonia when typing or buttoning), or appear at rest.9
Sensory Tricks (Alleviating Maneuvers or Geste Antagoniste). Initially considered a sign of psychogenicity, the sensory geste was later recognized as an important diagnostic clue for dystonia.10 The term refers to a voluntary maneuver that temporarily reduces the severity of dystonic postures or movements, usually consisting of a gentle touch. The sensory geste typically occurs in cervical dystonia, but may be present in other focal, generalized, or secondary forms of dystonia. In some cases, internal representation of the trick may also provide relief (eg, imagined interoceptive sensory tricks). In cervical dystonia, having the individual take the examiner’s hand and touch their chin may improve dystonia symptoms, although externally applied sensory tricks by the examiner are typically ineffective.11 Sensory tricks may not be recognized by individuals; therefore, their presence should always be investigated.
Mirror Dystonia. Mirror dystonia is defined as a unilateral posture or movement similar to a dystonic feature that can be elicited, usually on the more severely affected side, when contralateral movements or actions are performed. For example, in individuals with writer’s cramp, mirror dystonia may be evoked when they are writing with the unaffected limb while the affected hand rests on the desk, resulting in a dystonic posture in the dominant hand. This phenomenon may aid in selecting muscles for botulinum toxin injection, as it reveals the core triggering pattern of dystonia in the absence of compensatory movements.13
State Function. The term “state function” refers to a clinical phenomenon in which dystonia in a body part improves or worsens during a specific automatic motor activity. For example, focal lower limb dystonia or truncal dystonia may be triggered by walking forward but improved by walking backward or running. The mental representation of the movement that improves dystonia may itself result in an actual improvement of dystonia (ie, “imagined state function”).9
Null Point. The null point is the position in which the severity of the dystonic movement becomes less evident. The null point is helpful in case of dystonic movements with a tremulous quality. Examples are the lessening of tremor when turning the head to the side of the dystonic movements in cervical dystonia or the improvement of upper limb dystonia in certain positions.
Overflow. Overflow refers to unintentional muscle contraction that accompanies dystonia but is anatomically distinct from the primary dystonic movement commonly occurring at the peak of dystonic movements.4 Motor overflow may be seen in healthy individuals, but the threshold is lower in individuals with dystonia.12
Pathogenesis (Axis II)
Dystonia may be genetic or acquired.3 Given the shortcomings of the previous “DYT” classification system for genetic dystonias, a revised nomenclature has been established. According to this nomenclature, the prefix “DYT-” is followed by the name of the gene or the locus (eg, “DYT-TOR1A” instead of “DYT1”)13 (Table 1). The likelihood that dystonia has a genetic cause increases in combined forms, particularly when dystonia is associated with other neurologic abnormalities beyond movement disorders, or when symptom onset occurs before age 20 years.14 Acquired etiologies include perinatal brain injury, infection, drug or toxin exposure (Table 2), vascular insult, neoplastic disease, or autoimmune disorders. In most cases, the cause of adult-onset focal dystonia is unknown (formerly referred to as “idiopathic”).4

Pseudodystonia
First used by Fahn et al,2 the term “pseudodystonia” refers to “disorders that can mimic torsion dystonia, but are not generally considered to be a true dystonia.” Pseudodystonia has an identifiable cause and may be related to damage to the musculoskeletal system, peripheral nerve hyperexcitability, or compensatory abnormal postures (Table 3).9 The distinction between dystonia and pseudodystonia is of critical importance, as they have different diagnostic approaches, treatments, and prognosis.15 Pseudodystonia should be suspected when a posture is fixed or sustained, the onset is abrupt, and there is extreme pain out of proportion to the abnormal posture. The absence of sensory tricks is another clue. Functional dystonia has been included in the pseudodystonia category in the current updated classification of dystonia.4

Body Distribution
Dystonia is focal if it involves 1 body region (eg, blepharospasm, lower cranial dystonia, cervical dystonia, laryngeal dystonia, limb dystonia, truncal dystonia); segmental if it involves up to 3 adjacent body regions (eg, face and larynx, cervical region and upper limb); or multifocal if it involves up to 3 nonadjacent body regions (eg, one upper limb and the opposite lower limb). Hemidystonia refers to dystonia in 2 ipsilateral limbs, usually underpinned by a contralateral structural lesion. Generalized dystonia is more widespread than segmental or multifocal dystonia, regardless of involvement of the trunk.4
Blepharospasm is caused by abnormal contractions of the orbicularis oculi, procerus, and corrugator muscles. Lower cranial dystonia may involve the jaw, tongue, or pharynx. Jaw dystonia may cause abnormal jaw opening or closing or lateral jaw deviation. Laryngeal dystonia, or spasmodic dysphonia, includes an adductor type, which involves the thyroarytenoid muscles, producing a strangulated voice with breaks; and an abductor type, which involves the cricoarytenoid muscles, causing a breathy voice.
Dystonia in Isolation or Combined with Other Neurologic Disorders
Dystonia in the absence of other neurologic signs is called isolated dystonia. Dystonia is combined when accompanied by other movement disorders like tremor, myoclonus, chorea or ataxia; by other neurologic features (eg spasticity, lower motor neuron signs, seizures, cognitive decline); or by systemic manifestations, such as hepatic or cardiac abnormalities.4
Diagnostic Work-up
In individuals with adult-onset task-specific dystonia, no further diagnostic workup is needed. Brain MRI should be performed in all cases of early-onset dystonia, in combined forms, and when a lesion is suspected. Iron-sensitive sequences should be obtained to rule out neurodegeneration with brain iron accumulation (Figure 3). MRI can also be used to identify calcium accumulation (Figure 4), manganese accumulation (Figure 5), and structural causes such as pulvinar stroke (Figure 6).

Figure 3. MRI features of metal-related disorders (iron accumulation). The face of the giant panda sign (hyperintensity in the midbrain tegmentum with sparing of the red nuclei, substantia nigra, and tectum on the T2-weighted sequence) is pathognomonic of Wilson disease. A, The face of the miniature panda sign (hyperintensity in the pontine tegmentum with sparing of medial longitudinal fasciculi and central tegmental tracts) may also be present (ie, double panda sign). B, The eye of the tiger sign (hypointensity of bilateral globus pallidi with central hyperintensity on fluid-attenuated inversion recovery imaging) indicates pantothenate kinase–associated neurodegeneration (PANK-2). C, Hypointensity in bilateral globus pallidi with hyperintense streaks (arrowheads) at bilateral medial medullary lamina on the T2-weighted sequence indicates mitochondrial membrane protein–associated neurodegeneration (MPAN). D, Hyperintense signal with central linear hypointensity in bilateral substantia nigra on T1-weighted imaging reveals β-propeller protein–associated neurodegeneration (BPAN).

Figure 4. CT and MRI features of calcium accumulation in Fahr disease. On CT scans, primary familial basal ganglia calcification is observed bilaterally in the dentate nuclei, cerebellar white matter, basal ganglia, and thalami, and diffusely across the cerebral cortices (A, B). A corresponding susceptibility-weighted MRI scan shows hypointensity in the bilateral basal ganglia and thalami (C).

Figure 5. MRI features of manganese accumulation. Hyperintensity on T1-weighted sequences in bilateral basal ganglia, especially the globus pallidus, is typical of manganese accumulation (eg, due to secondary hepatic failure, acquired hepatocerebral degeneration, manganese toxicity, genetic defects in the manganese transporter gene).
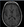
Figure 6. MRI features of pulvinar stroke. Right thalamic hypointensity is seen on the T1-weighted image.
Laboratory studies should include evaluation of liver and renal function, as well as copper, ceruloplasmin, 24-hour urinary copper excretion, manganese, calcium, and parathyroid hormone levels.16 If a genetic cause is suspected, a single gene test or a dystonia panel may be performed, and in case of strong suspicion, whole-genome sequencing or genome-
wide sequencing should be performed as a second step (see Figures 1, 2). If the diagnostic investigation does not reveal an etiology, the dystonia is classified as unknown.
Treatment
Treatment of dystonia depends on the etiology and the body distribution, and whether pathogenesis-targeted therapies are available. Targeted therapies are available for some genetic dystonias.5
Multiple symptomatic treatments are available for dystonia (Table 4). The goal of symptomatic treatments is to provide relief from associated pain and discomfort and to prevent contractures or other orthopedic complications of untreated abnormal postures. The gold standard treatment for most forms of focal dystonia is botulinum toxin injection. Generalized forms of dystonia may benefit from oral therapies, and, in the most disabling cases, from deep brain stimulation (DBS), most commonly targeting the globus pallidus internus. Genetic profile is an independent factor affecting response to DBS.17

Oral therapies include trihexyphenidyl, an anticholinergic drug; baclofen, a muscle relaxant, administered orally or with an intrathecal pump, particularly for individuals with associated generalized spasticity or painful dystonia; and clonazepam, mainly as an add-on therapy. In individuals with tardive dystonia, therapeutic options include discontinuation of the offending agent or switching to clozapine; agents such as vesicular monoamine transport 2 inhibitors, amantadine, or trihexyphenidyl may be helpful. A considerable minority of individuals with myoclonus-dystonia and laryngeal dystonia report sensitivity to alcohol and its analogue, sodium oxybate, a salt of gamma-hydroxybutyrate.18
Avoidance of triggers and treatment with carbamazepine/oxcarbazepine is the therapy of choice for paroxysmal kinesigenic dystonia. Clonazepam is helpful in nonkinesigenic forms. Rehabilitation therapy, in addition to medical therapy, may also contribute to improved function in specific forms of dystonia. Status dystonicus, which is characterized by severe, generalized, and continuous dystonia, is seen more frequently in children with neurodevelopmental disorders, and rarely in individuals with DBS due to hardware failure. Status dystonicus requires urgent hospital admission and, in some cases, management in an intensive care unit with sedative infusions or DBS.19
Non-motor symptoms, including mood disorders, pain, fatigue, and sleep disruption, are increasingly recognized as major contributors to reduced quality of life in dystonia. Improvement of anxiety, depression, and pain has been reported following Botulinum toxin injections. Systematic assessment of these symptoms is fundamental to a multifaceted treatment approach that combines physical therapy, psychotherapy, and pharmacological interventions.21
Conclusion
Phenomenology is key to the diagnosis of dystonia, which should be distinguished from its mimics. Clinical examination provides essential information to determine the pathogenesis and guide the diagnostic workup. A prompt and accurate diagnosis is crucial to guide the therapeutic approach.
Ready to Claim Your Credits?
You have attempts to pass this post-test. Take your time and review carefully before submitting.
Good luck!
Recommended
- Movement Disorders
Bill Nye on Ataxia, Family Risk, and the Search for Answers
Bill Nye; Lauren Moore, PhDBill Nye; Lauren Moore, PhD - Imaging & Testing
Neuroimaging: Clinical Impact of Imaging Findings
Laszlo Mechtler, MD, FAAN, FEAN, FASN, FAHSLaszlo Mechtler, MD, FAAN, FEAN, FASN, FAHS

