Neuromuscular Notes: Genetics of Amyotrophic Lateral Sclerosis

Amyotrophic lateral sclerosis (ALS) is a progressive disorder that affects motor neurons, leading to ultimate paralysis and respiratory failure. Although the majority of ALS cases are sporadic (SALS), familial ALS (FALS) accounts for approximately 10% of the population with ALS. As our understanding of ALS phenotypes and disease mechanisms has grown, so has the pace of ALS gene discovery. This research has paved the way for trials in ALS gene therapy, offering hope for people affected by FALS. The neurology community has the opportunity to educate individuals at risk for the inherited forms of ALS and provide information on evolving experimental gene therapy programs.
Advances in genetic testing techniques and research initiatives focused on ALS gene discovery have led to an explosion in the number of known ALS causative genes and potential modifiers over the last decade. Since the discovery of the SOD1 gene,1 which is the most common ALS causative gene accounting for 20% of FALS, more than 50 genes have been associated with ALS (Figure).2 The majority of mutations are dominantly inherited with high penetrance and account for a small portion of apparent SALS (ie, 1%-3% of SALS are secondary to SOD1 mutations, whereas 5% of SALS can be attributed to the C9orf72 repeat expansion). For the treating neurologist, it may be useful to conceptualize the most common ALS causative genes in the context of phenotypic presentation (Table).
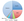

Amyotrophic Lateral Sclerosis Gene Variants
SOD1 Variants
The SOD1 gene encodes a ubiquitously expressed cytoplasmic enzyme, Cu-Zn superoxide dismutase, which has an antioxidant function. When mutated, SOD1 has a complex toxicity. More than 150 ALS-causing variants have been identified in nearly every region of the SOD1 polypeptide.2 Most SOD1 variants lead to lower motor neuron-predominant phenotypes.3 The rate of progression is dependent on the variant harbored by an individual, ranging from rapidly progressive SOD1A4V4 to variants associated with a more indolent course, such as SOD1D90A.3
C9orf72 Repeat Expansion
Pathogenic expansion of GGGGCC repeats in C9orf72 has been detected on chromosome 9p21.5 It is proposed that this causes disease via gain of function leading to accumulation of RNA foci and RNA-binding proteins or toxicity from dipeptide repeat protein translation and impaired nucleocytoplasmic transport.2,6 Healthy individuals harbor between 2 and 23 repeats. In contrast, individuals with ALS typically have hundreds to thousands of repeats. The repeat expansion is inherited in an autosomal dominant manner and represents the most common form of familial ALS (25%), with a higher prevalence in Scandinavian populations. The related phenotype is representative of the entire clinical spectrum of ALS.7 Importantly, these individuals have a higher incidence of bulbar-onset ALS and ALS with parkinsonism, and there is a higher coincidence of the behavioral variant of frontotemporal dementia and family history of dementia. The penetrance (ie, proportion of gene carriers expressing the associated phenotype) of the disease appears to increase with age, with 99.5% penetrance by age 83.8 Cohort-based studies provide potential evidence for anticipation (ie, earlier appearance of a genetic disease with each succeeding generation) in C9orf72 (C9)-related ALS, with an unclear relation to expansion size.7 Rates of survival for people with C9-related ALS also appear shorter than for those without the repeat expansion.9 These findings require verification in larger cohorts but are a step toward understanding the broad spectrum of C9-related disease.
KIF5A-Associated Amyotrophic Lateral Sclerosis
Mutations in the C-terminus of the kinesin family member 5A (KIF5A) gene are also associated with ALS.10,11 People with this loss-of-function mutation have a distinctive ALS phenotype with a younger disease onset (median age of onset 46.5) and slower disease course (median survival of approximately 10 years) compared to the average person with ALS.12 Mutations in the N-terminus of this same gene can cause disorders, such as hereditary spastic paraplegia (SPG10) and Charcot–Marie–Tooth disease type 2. The association of the KIF5A mutation with ALS exemplifies how large-scale genetic discovery research initiatives in ALS can broaden our understanding of inherited neurodegenerative disease to help guide patients and their families regarding prognosis and genetic risk.
Other Variants
There is enormous interest in genetic variants that could potentially modify ALS susceptibility, clinical phenotype, or survival. Single nucleotide variants (SNVs) in the UNC13A gene are associated with population-specific ALS risk in a cohort from the United Kingdom and different SNVs correlated with long or short survival.13 In people with ALS, EPHA4 expression inversely correlates with disease onset and survival, whereas in contrast, EPHA4 loss-of-function mutations are associated with longer survival.14 Finally, a variant of TREM2, which has been found to play a role in microglial activation, is a risk factor for sporadic ALS.15 Genetic modifiers with an impact on survival could be helpful in prognostication or targeted directly for therapeutic intervention.
Using Amyotrophic Lateral Sclerosis Genetics
The field of neurogenetics can be challenging to navigate because of the rapid pace of genetic discovery and growing selection of available tests. Genetic testing can be complex and collaboration with a genetic counselor can both help to delineate the appropriate testing and provide support to patients and providers. If a genetic counselor is not available, referral to a specialized neuromuscular disease center may be appropriate.
Targeted single-gene testing may first be sought if the genetic variant in the family is known. In familial cases where a gene has not yet been identified, next generation sequencing (NGS) panels now allow for timely and simultaneous evaluation for multiple ALS-causative genes. If targeted variant tests or NGS panels are unrevealing, high-throughput methods such as whole exome sequencing (WES), which is analysis of the entire coding portion of the genome, or whole genome sequencing (WGS), which spans the entire genome, can be used for more in-depth analyses.
When to Test Genetics
In the symptomatic person with ALS, genetic testing should be strongly considered if there is a family history of ALS, dementia is present, or onset occurred before age 50. The patient’s rationale for learning their gene status should be sought. Knowledge of genetic risk can directly affect reproductive decisions for the person affected, their spouse, and/or subsequent generations. In the setting of early symptoms concerning for ALS and a family history of ALS or dementia, positive genetic testing can expedite an otherwise often lengthy diagnostic process. Before testing, it is important to explore the individual’s context (eg, social support and mental health stability), recognizing that genetic test results may cause added psychologic stress.
Utility of Genetic Testing
A diagnosis of FALS can expand options for experimental therapy targeting an individual’s own gene mutation—a unique opportunity in a field where precise etiology of disease is often unknown. If clinical trials in FALS are not locally available, the caring neurologist could search clinicaltrials.gov or ALS-related clinical trials websites and direct individuals accordingly.
A Time of Great Promise
Researchers are now partnering with families affected by FALS in presymptomatic gene carrier natural history studies, striving to identify the earliest biological changes of ALS at the point of symptom conversion. In this exciting time of genetic discovery and promising gene therapy, there is finally hope for effective treatments and ultimately disease prevention for people affected by inherited forms of ALS.
Disclosure
KN has received funding from the ALS Association, American Academy of Neurology, ALS Finding a Cure, Target ALS, the Muscular Dystrophy Association, Brainstorm Therapeutics, and the Salah Foundation, provided consulting for Avanir Pharmaceuticals and completed the Anne B. Young Fellowship in Therapeutic Development at Massachusetts General Hospital and Biogen.
Ready to Claim Your Credits?
You have attempts to pass this post-test. Take your time and review carefully before submitting.
Good luck!
Recommended
- Neuromuscular
Patient-Reported Outcome Measures in Neuromuscular Disorders
Brianne Kelly, MD; Saba Nawaz, MDBrianne Kelly, MD; Saba Nawaz, MD

